The Source Code of Life: Using Python to Explore Our DNA¶
 Researchers just found the gene responsible for mistakenly thinking we've found the gene for specific things. It's the region between the start and the end of every chromosome, plus a few segments in our mitochondria.
Researchers just found the gene responsible for mistakenly thinking we've found the gene for specific things. It's the region between the start and the end of every chromosome, plus a few segments in our mitochondria.
Bioinformatics¶
HINT: If you're viewing this notebook as slides, press the "s" key to see a bunch of extra notes.
 Image: http://www.sciencemag.org/sites/default/files/styles/article_main_large/public/images/13%20June%202014.jpg?itok=DPBy5nLZ
Image: http://www.sciencemag.org/sites/default/files/styles/article_main_large/public/images/13%20June%202014.jpg?itok=DPBy5nLZ
What is DNA?¶
A long string of A, C, G, and T (bases)
ACGTTCATGG <- Ten bases
ACGTTCATGGATGTGACCAG <- Twenty bases
etc...Well, not really a string...
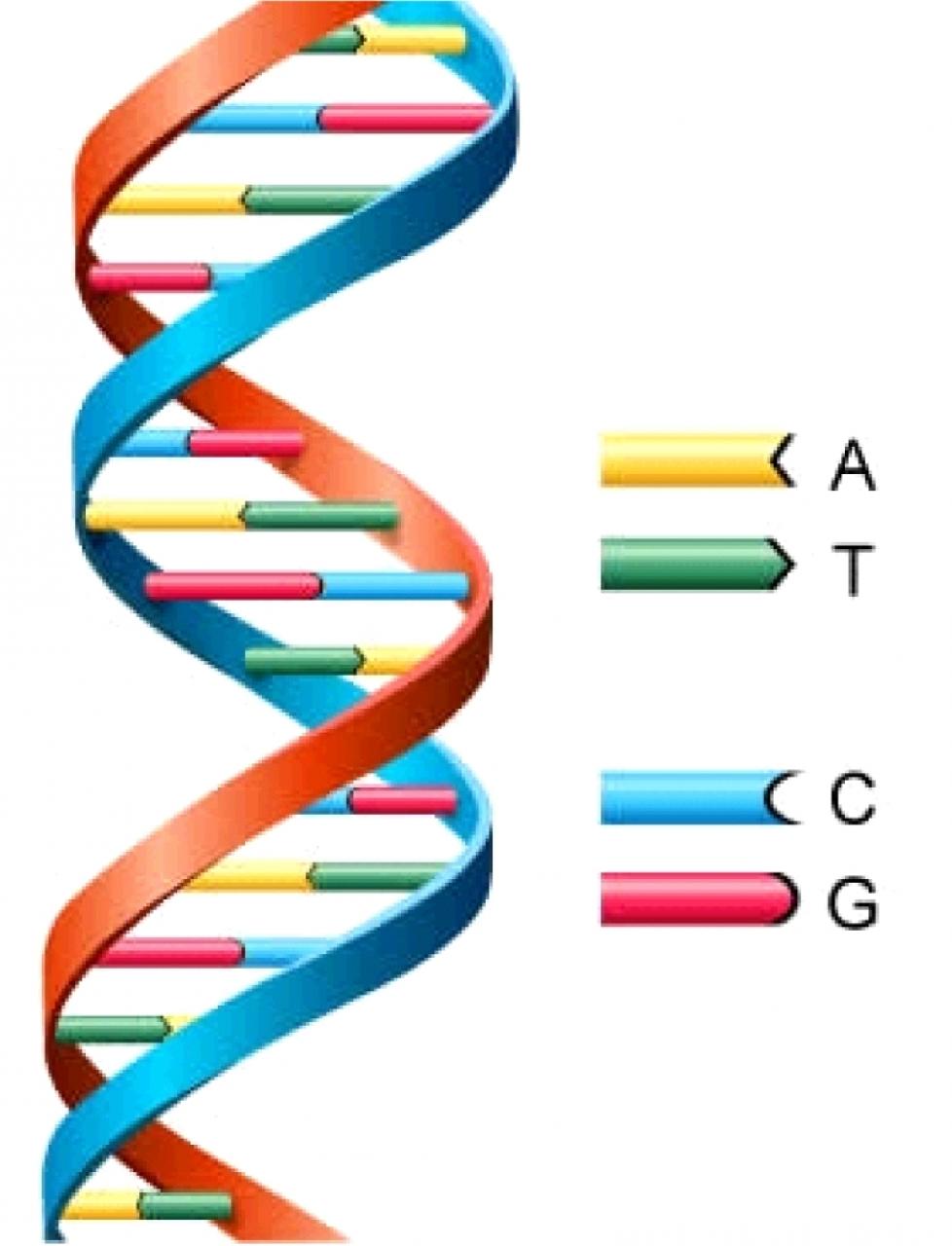 Two strings stuck together? But if you know one you know the other...
Two strings stuck together? But if you know one you know the other...
The Human Genome¶
It's big. How big?
About 3,200,000,000bp (bp is base pairs). Actual file format commonly used in bioinformatics (FASTA):
>Sequence0
TTTCTGACTAACACTACAATTACCACTTGATGTTACCGACTAAGTGGTACGACTTGCTAGAACCGACTCTCGTACGTAT
CGCAGACTAGTGCGCGCGCTTAGTGACTATACTAGAATATACCTGGGGCCCAAGGAGTGTCGGGCGATCGTCCTTGAAA
TAAATATCTCAACCATCGTCATCTAGGGGGAACAGAGCGGTGGGCAGGTCCCAACCTGTTTATTTGTGTTGCTAACACT
ACGGCGCAGCTGCTCAAGTAGGTGCGATTATCGAGTAGAGGCTCCACCGGCTCTATGTGCCACGCATCTACTGAACCGA
ATTCTATCCCTGATACTCCAGAAGGTCGCAGGTTTACAGACACGTTTCAGCTCGAGAGGCCATCGATTATCTTAATATA
CCACACTGCCGAATAGCATGCCCGTAGAATCCAAGCCACGAGATAGCGTTACTTAATGAGTACCCAACGCAAATGAGGT
TGATTATCCCTAACCTGCAATCTAGGCCTTGTTCTGGAGGGGGTTATCCTTTATAGTTGATTACTTACACTCACCATGT
TCGTAGTCGGAACTCACCGATTAAGACCGATTTTACTATGGGAAGGCCAGGTTACACCTGTTTCGGGGGGGCCGCGGCG
GGTTACTTTAACCTGTCCATCCATCAGTCACTGGGCGCCAAGATTCTCCTATAGTTATATCCGCCCTTTGATTTAAACC
TAGGCCTACCTCAACGAACTGGGCCATGGGGTTCACACAGAAACAAGGGGGATAGACAGTCTTATTGAGCGCTTCTGAA
CAGCGTGTGTTCACGGTACGGCAATACCACCAGTAAACCGAGAACAGTGTTGAAGGTGATCGAACACGTGTTTTCTTCA
CCGTAGGGCTTCTAGGGAGTATCGCCCCCATATAGGCAGACGAGAAGGACTGTCACGCGCGGAGATCGATAATACGTAT
AACACAAGCACAGTAACTGCCCCGACCGGCTAAAGGACGTGGCCCAGTGTACCCAACGTACGTAATTGCAAGAGGTCTG
TCTGTCATCCCGAGGACTGCTTCTATAACTCGTTGAGGGCACTAGGCTTGAGACAATCAGCTTCGCTCGTCACGATTTT
ACTTTTTTCCTGGAAAAGCCCCCCCACAGACTATCAGGTCGCGCTTACCATACCAGTCCTTCTTGATAAGCCAATCCGT
ATTAGGTAGATTAAGCTGACAGTCGGGGCGACTCTTTGGAAACAGTATTCCCGTTTCGGGCACCTAGGATTCAGGCTTG
TACAACGATCATAGACGTCGCGGAAAGAAATAGCACAGTGTAGGAGCTGGTCGTGACCCGTGCTGTCAAGTTTATTGCA
CGGCTTGCTAAAAGGTACAGTGTAACGTTTCACAAACAAGCGAGACCCATTGTTGGTCTAACGCTATCGTACTTGATAC
CAGCCTGTGACGTCACGCGAAATCGTCTGTATAACTAGTTCTTCCCCGACTGCCACGGTATCCCAAAATTACATACTGA
CAGGACCTCTTCCATATTCATCAGGACTCGACGAAGCGCGCCCCGTGTAGTACGCGAAAATTATACCGTCCGTAGGTACNow picture this 2 million more times.
{kind=link}
Sequencing DNA¶
YouTubeVideo('fCd6B5HRaZ8', start=135)
Sequence Alignment¶
Now things start to get fun
That's a nice-looking lake...

Until now!

First step: Assign scores¶
If a base matches with itself, it gets a score of one. Otherwise it gets a score of zero.
| A | C | G | T | |
|---|---|---|---|---|
| A | 1 | 0 | 0 | 0 |
| C | 0 | 1 | 0 | 0 |
| G | 0 | 0 | 1 | 0 |
| T | 0 | 0 | 0 | 1 |
So these two sequences:
AACTGTGGTAC
ACTTGTGGAAC
10011111011have an alignment score of eight.
What about gaps?¶
Yes, unfortunately there are going to be gaps.
We'll add a "gap penalty" of -3.
So the score for this alignment:
ACTACA-ACGTTGAC
A-TAGAAACGCT-AC
1 1101 11101 11
-3 -3 -3is just one
Scoring matrix¶
| - | A | C | G | T | T | T | G | T | C | G | C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | -3 | -6 | -9 | -12 | -15 | -18 | -21 | -24 | -27 | -30 | -33 |
| A | -3 | |||||||||||
| C | -6 | |||||||||||
| T | -9 | |||||||||||
| T | -12 | |||||||||||
| T | -15 | |||||||||||
| C | -18 | |||||||||||
| T | -21 | |||||||||||
| G | -24 | |||||||||||
| C | -27 |
$$ S_{m,n}=\left\{ \begin{array}{ll} S_{m-1,n} + gap\\ S_{m,n-1} + gap\\ S_{m-1,n-1} + B(a,b)\\ \end{array} \right. $$
| - | A | C | G | T | T | T | G | T | C | G | C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | -3 | -6 | -9 | -12 | -15 | -18 | -21 | -24 | -27 | -30 | -33 |
| A | -3 | 1 | -2 | -5 | -8 | -11 | -14 | -17 | -20 | -23 | -26 | -29 |
| C | -6 | |||||||||||
| T | -9 | |||||||||||
| T | -12 | |||||||||||
| T | -15 | |||||||||||
| C | -18 | |||||||||||
| T | -21 | |||||||||||
| G | -24 | |||||||||||
| C | -27 |
We need to keep track of where that score came from.
| - | A | C | G | T | T | T | G | T | C | G | C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | ←-3 | ←-6 | ←-9 | ←-12 | ←-15 | ←-18 | ←-21 | ←-24 | ←-27 | ←-30 | ←-33 |
| A | ↑-3 | ↖1 | ←-2 | ←-5 | ←-8 | ←-11 | ←-14 | ←-17 | ←-20 | ←-23 | ←-26 | ←-29 |
| C | ↑-6 | |||||||||||
| T | ↑-9 | |||||||||||
| T | ↑-12 | |||||||||||
| T | ↑-15 | |||||||||||
| C | ↑-18 | |||||||||||
| T | ↑-21 | |||||||||||
| G | ↑-24 | |||||||||||
| C | ↑-27 |
| - | A | C | G | T | T | T | G | T | C | G | C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | ←-3 | ←-6 | ←-9 | ←-12 | ←-15 | ←-18 | ←-21 | ←-24 | ←-27 | ←-30 | ←-33 |
| A | ↑-3 | ↖1 | ←-2 | ←-5 | ←-8 | ←-11 | ←-14 | ←-17 | ←-20 | ←-23 | ←-26 | ←-29 |
| C | ↑-6 | ↑-2 | ↖2 | ←1 | ←-4 | ←-7 | ←-10 | ←-13 | ←-16 | ↖-19 | ←-22 | ↖-25 |
| T | ↑-9 | ↑-5 | ↑-1 | ↖2 | ↖0 | ←-3 | ←-6 | ←-9 | ←-12 | ←-15 | ←-18 | ←-21 |
| T | ↑-12 | ↑-8 | ↑-4 | ↑-1 | ↖3 | ↖1 | ←-2 | ←-5 | ↖-8 | ←-11 | ←-14 | ←-17 |
| T | ↑-15 | ↑-11 | ↑-7 | ↖-4 | ↑0 | ↖4 | ↖2 | ←-1 | ↖-4 | ←-7 | ←-10 | ←-13 |
| C | ↑-18 | ↑-14 | ↖-10 | ↖-7 | ↑-3 | ↑1 | ↖4 | ↖2 | ↖-1 | ↖-3 | ←-6 | ←-9 |
| T | ↑-21 | ↑-17 | ↑-13 | ↑-10 | ↑-6 | ↑-2 | ↖2 | ↖4 | ↖3 | ←0 | ↖-3 | ←-6 |
| G | ↑-24 | ↑-20 | ↑-16 | ↖-12 | ↑-9 | ↑-5 | ↑-1 | ↖3 | ↖4 | ↖3 | ↖1 | ←-2 |
| C | ↑-27 | ↑-23 | ↖-19 | ↑-15 | ↖-12 | ↑-8 | ↑-4 | ↑0 | ↖3 | ↖5 | ↖3 | ↖2 |
Now just follow the arrows from that bottom-right corner back to the top-left zero.
ACGTTTGTCGC
|| ||| | ||
AC-TTTCT-GCLet's code this!¶
See Python for Bioinformatics for the inspiration for this demo.
Here is the "substitution matrix" and its corresponding "alphabet":
dna_sub_mat = np.array(
[[ 1, 0, 0, 0],
[ 0, 1, 0, 0],
[ 0, 0, 1, 0],
[ 0, 0, 0, 1]])
dbet = 'ACGT'
And here are we calculate the scores and arrows as separate matrices
def nw_alignment(sub_mat, abet, seq1, seq2, gap=-8):
# Get the lengths of the sequences
seq1_len, seq2_len = len(seq1), len(seq2)
# Create the scoring and arrow matrices
score_mat = np.zeros((seq1_len+1, seq2_len+1), int)
arrow_mat = np.zeros((seq1_len+1, seq2_len+1), int)
# Fill first column and row of score matrix with scores based on gap penalty
score_mat[0] = np.arange(seq2_len+1) * gap
score_mat[:,0] = np.arange(seq1_len+1) * gap
# Fill top row of arrow matrix with ones (left arrow)
arrow_mat[0] = np.ones(seq2_len+1)
for seq1_pos, seq1_letter in enumerate(seq1):
for seq2_pos, seq2_letter in enumerate(seq2):
f = np.zeros(3)
# Cell above + gap penalty
f[0] = score_mat[seq1_pos,seq2_pos+1] + gap
# Cell to left + gap penalty
f[1] = score_mat[seq1_pos+1,seq2_pos] + gap
n1 = abet.index(seq1_letter)
n2 = abet.index(seq2_letter)
# Cell to upper-left + alignment score
f[2] = score_mat[seq1_pos,seq2_pos] + sub_mat[n1,n2]
score_mat[seq1_pos+1, seq2_pos+1] = f.max()
arrow_mat[seq1_pos+1, seq2_pos+1] = f.argmax()
return score_mat, arrow_mat
What does our result look like?
s1 = 'ACTTCTGC'
s2 = 'ACGTTTGTCGC'
score_mat, arrow_mat = nw_alignment(dna_sub_mat, dbet, s1, s2, gap=-3)
print(score_mat)
print(arrow_mat)
Now we need a way to get the sequences back out of our scoring matrix:
def backtrace(arrow_mat, seq1, seq2):
align1, align2 = '', ''
align1_pos, align2_pos = arrow_mat.shape
align1_pos -= 1
align2_pos -= 1
selected = []
while True:
selected.append((align1_pos, align2_pos))
if arrow_mat[align1_pos, align2_pos] == 0:
# Up arrow, add gap to align2
align1 += seq1[align1_pos-1]
align2 += '-'
align1_pos -= 1
elif arrow_mat[align1_pos, align2_pos] == 1:
# Left arrow, add gap to align2
align1 += '-'
align2 += seq2[align2_pos-1]
align2_pos -= 1
elif arrow_mat[align1_pos, align2_pos] == 2:
# Up-arrow arrow, no gap
align1 += seq1[align1_pos-1]
align2 += seq2[align2_pos-1]
align1_pos -= 1
align2_pos -= 1
if align1_pos==0 and align2_pos==0:
break
# reverse the strings
return align1[::-1], align2[::-1], selected
a1, a2, selected = backtrace(arrow_mat, s1, s2)
print(a1)
print(a2)
Sometimes it's nice to see the scoring matrix, though, so here's a function to visualize it
def visual_scoring_matrix(seq1, seq2, score_mat, arrow_mat):
visual_mat = []
for i, row in enumerate(arrow_mat):
visual_mat_row = []
for j, col in enumerate(row):
if col == 0:
arrow = '↑'
elif col == 1:
arrow = '←'
else:
arrow = '↖'
visual_mat_row.append(arrow + ' ' + str(score_mat[i,j]))
visual_mat.append(visual_mat_row)
visual_mat = np.array(visual_mat, object)
tab = plt.table(
cellText=visual_mat,
rowLabels=['-'] + list(s1),
colLabels=['-'] + list(s2),
loc='center')
tab.scale(2, 3)
tab.set_fontsize(30)
plt.axis('tight')
plt.axis('off')
align1, align2, selected = backtrace(arrow_mat, seq1, seq2)
for pos in selected:
y, x = pos
tab._cells[(y+1, x)]._text.set_color('green')
tab._cells[(y+1, x)]._text.set_weight(1000)
plt.show()
visual_scoring_matrix(s1, s2, score_mat, arrow_mat)

Let's generate some sequences and see how fast this is
def random_dna_seq(length=100):
seq = [random.choice(dbet) for x in range(length)]
return ''.join(seq)
def mutate_dna_seq(seq, chance=1/5):
mut_seq_base = [random.choice(dbet) if random.random() < chance else x for x in seq]
mut_seq_indel = [random.choice(('', x + random.choice(dbet))) if random.random() < chance else x for x in mut_seq_base]
return ''.join(mut_seq_indel)
s1 = random_dna_seq()
s2 = mutate_dna_seq(s1)
print(s1[:25], len(s1))
print(s2[:25], len(s2))
a = %timeit -o nw_alignment(dna_sub_mat, dbet, s1, s2, gap=-3)
print('{:.1f} years for the whole genome'.format(a.best * 2300000000 / 60 / 60 / 24 / 365.25))
Let's make it faster! (Just because)
| - | A | C | G | T | T | T | G | T | C | G | C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0 | ←-3 | ←-6 | ←-9 | ←-12 | ←-15 | ←-18 | ←-21 | ←-24 | ←-27 | ←-30 | ←-33 |
| A | ↑-3 | ↖1 | ←-2 | |||||||||
| C | ↑-6 | ↑-2 | ||||||||||
| T | ↑-9 | |||||||||||
| T | ↑-12 | |||||||||||
| T | ↑-15 | |||||||||||
| C | ↑-18 | |||||||||||
| T | ↑-21 | |||||||||||
| G | ↑-24 | |||||||||||
| C | ↑-27 |
We can't calculate a whole row or column at a time because the values depend on those in the same row/column. But what about diagonals?
We'll "get rid of" our nested loop (really just abstract it into a faster numpy "loop")
This is going to take a couple more steps, but it will be worth it in the end.
First we pre-calculate the "upper-left score" for each location.
def sub_values(sub_mat, abet, seq1, seq2):
# convert the sequences to numbers
seq1_ind = [abet.index(i) for i in seq1]
seq2_ind = [abet.index(i) for i in seq2]
sub_vals = np.array([[0] * (len(seq2)+1)] + [[0] + [sub_mat[y, x] for x in seq2_ind] for y in seq1_ind], int)
return sub_vals
sub_values(dna_sub_mat, dbet, 'AACGTTA', 'AAGCTTAAAAAAAA')
Then we get a list of all the diagonals in the matrix.
def diags(l1, l2):
ys = np.array([np.arange(l1) + 1 for i in np.arange(l2)])
xs = np.array([np.arange(l2) + 1 for i in np.arange(l1)])
diag_ys = [np.flip(ys.diagonal(i), 0) for i in range(1-l2, l1)]
diag_xs = [xs.diagonal(i) for i in range(1-l1, l2)]
index_list = []
for y, x in zip(diag_ys, diag_xs):
index_list.append([y, x])
return index_list
diags(6, 3)
And here's the actual function. It takes the same arguments and produces the same matrices.
def FastNW(sub_mat, abet, seq1, seq2, gap=-8):
sub_vals = sub_values(sub_mat, abet, seq1, seq2)
# Get the lengths of the sequences
seq1_len, seq2_len = len(seq1), len(seq2)
# Create the scoring and arrow matrices
score_mat = np.zeros((seq1_len+1, seq2_len+1), int)
arrow_mat = np.zeros((seq1_len+1, seq2_len+1), int)
# Fill first column and row of score matrix with scores based on gap penalty
score_mat[0] = np.arange(seq2_len+1) * gap
score_mat[:,0] = np.arange(seq1_len+1) * gap
# Fill top row of arrow matrix with ones (left arrow)
arrow_mat[0] = np.ones(seq2_len+1)
# Get the list of diagonals
diag_list = diags(seq1_len, seq2_len)
# fill in the matrix
for diag in diag_list:
# Matrix to hold all three possible scores for every element in the diagonal
f = np.zeros((3, len(diag[0])), float)
# Cell above + gap penalty for every cell in the diagonal
x, y = diag[0]-1, diag[1]
f[0] = score_mat[x, y] + gap
# Cell to the left + gap penalty for every cell in the diagonal
x, y = diag[0], diag[1]-1
f[1] = score_mat[x, y] + gap
# Cell to the upper left + alignment score for every cell in the diagonal
x, y = diag[0]-1, diag[1]-1
f[2] = score_mat[x,y] + sub_vals[diag]
max_score = (f.max(0))
max_score_pos = f.argmax(0)
score_mat[diag] = max_score
arrow_mat[diag] = max_score_pos
return score_mat, arrow_mat
So how much faster is it?
s1 = random_dna_seq()
s2 = mutate_dna_seq(s1)
a = %timeit -o nw_alignment(dna_sub_mat, dbet, s1, s2)
print('{:.1f} years for the whole genome'.format(a.best * 2300000000 / 60 / 60 / 24 / 365.25))
a = %timeit -o FastNW(dna_sub_mat, dbet, s1, s2)
print('{:.1f} years for the whole genome'.format(a.best * 2300000000 / 60 / 60 / 24 / 365.25))
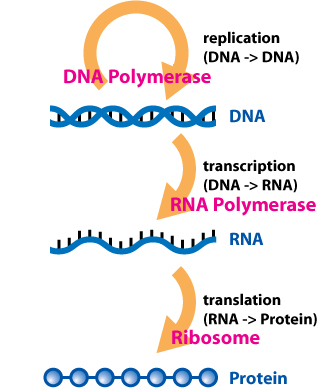
Now why did we use the substitution matrix?¶
Here's how DNA translates to protein:
So we can align proteins, too!
blosum50 = np.array(
[[ 5,-2,-1,-2,-1,-1,-1, 0,-2,-1,-2,-1,-1,-3,-1, 1, 0,-3,-2, 0],
[-2, 7,-1,-2,-1, 1, 0,-3, 0,-4,-3, 3,-2,-3,-3,-1,-1,-3,-1,-3],
[-1,-1, 7, 2,-2, 0, 0, 0, 1,-3,-4,-0,-2,-4,-2,-1, 0,-4,-2,-3],
[-2,-2, 2, 8,-4, 0, 2,-1,-1,-4,-4,-1,-4,-5,-1, 0,-1,-5,-3,-4],
[-1,-4,-2,-4,13,-3,-3,-3,-3,-2,-2,-3,-2,-2,-4,-1,-1,-5,-3,-1],
[-1,-1, 0, 0,-3, 7, 2,-2, 1,-3,-2, 2, 0,-4,-1,-0,-1,-1,-1,-3],
[-1, 0, 0, 2,-3, 2, 6,-3, 0,-4,-3, 1,-2,-3,-1,-1,-1,-3,-2,-3],
[ 0,-3, 0,-1,-3,-2,-3, 8,-2,-4,-4,-2,-3,-4,-2, 0,-2,-3,-3,-4],
[-2, 0, 1,-1,-3, 1, 0,-2,10,-4,-3, 0,-1,-1,-2,-1,-2,-3,-1, 4],
[-1,-4,-3,-4,-2,-3,-4,-4,-4, 5, 2,-3, 2, 0,-3,-3,-1,-3,-1, 4],
[-2,-3,-4,-4,-2,-2,-3,-4,-3, 2, 5,-3, 3, 1,-4,-3,-1,-2,-1, 1],
[-1, 3, 0,-1,-3, 2, 1,-2, 0,-3,-3, 6,-2,-4,-1, 0,-1,-3,-2,-3],
[-1,-2,-2,-4,-2, 0,-2,-3,-1, 2, 3,-2, 7, 0,-3,-2,-1,-1, 0, 1],
[-3,-3,-4,-5,-2,-4,-3,-4,-1, 0, 1,-4, 0, 8,-4,-3,-2, 1, 4,-1],
[-1,-3,-2,-1,-4,-1,-1,-2,-2,-3,-4,-1,-3,-4,10,-1,-1,-4,-3,-3],
[ 1,-1, 1, 0,-1, 0,-1, 0,-1,-3,-3, 0,-2,-3,-1, 5, 2,-4,-2,-2],
[ 0,-1, 0,-1,-1,-1,-1,-2,-2,-1,-1,-1,-1,-2,-1, 2, 5,-3,-2, 0],
[-3,-3,-4,-5,-5,-1,-3,-3,-3,-3,-2,-3,-1, 1,-4,-4,-3,15, 2,-3],
[-2,-1,-2,-3,-3,-1,-2,-3, 2,-1,-1,-2, 0, 4,-3,-2,-2, 2, 8,-1],
[ 0,-3,-3,-4,-1,-3,-3,-4,-4, 4, 1,-3, 1,-1,-3,-2, 0,-3,-1, 5]])
pbet = 'ARNDCQEGHILKMFPSTWYV'
s1 = [random.choice(pbet) for _ in range(10)]
s2 = [random.choice(pbet) if random.random() < .25 else x for x in s1] + [random.choice(pbet) for _ in range(10)]
score_mat, arrow_mat = FastNW(blosum50, pbet, s1, s2)
visual_scoring_matrix(s1, s2, score_mat, arrow_mat)

Other things we can do with this algorithm:¶
- Local alignment
- Affine gap penalties
Other useful bioinformatics Python packages:¶
from pysam import FastaFile
from os.path import getsize
print(getsize('Homo_sapiens.GRCh38.dna.primary_assembly.fasta.gz')/1024**2, 'MiB')
with FastaFile('Homo_sapiens.GRCh38.dna.primary_assembly.fasta.gz') as myfasta:
chr17len = myfasta.get_reference_length('17')
print(chr17len, 'bp')
seq = myfasta.fetch('17', int(chr17len/2), int(chr17len/2)+500)
print(seq)
with FastaFile('Homo_sapiens.GRCh38.dna.primary_assembly.fasta.gz') as myfasta:
chr17len = myfasta.get_reference_length('17')
%timeit myfasta.fetch('17', int(chr17len/2), int(chr17len/2)+500)
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
coding_dna = Seq("ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG", IUPAC.unambiguous_dna)
print(coding_dna.__repr__())
print(coding_dna.translate().__repr__())
Any questions?¶
Other possible topics:¶
How do you actually get from reads to something usable?
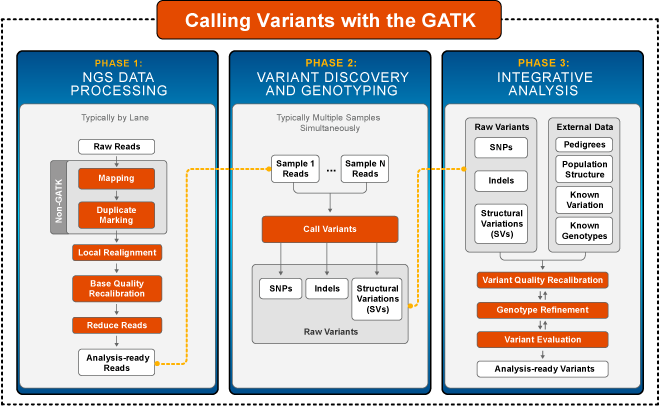
Why does bioinformatics use flat files and not databases for everything? The best answer I can give is that of Heng Li when talking about Tabix:
It is straightforward to achieve overlap queries using the standard B-tree index (with or without binning) implemented in all SQL databases, or the R-tree index in PostgreSQL and Oracle. But there are still many reasons to use tabix. Firstly, tabix directly works with a lot of widely used TAB-delimited formats such as GFF/GTF and BED. We do not need to design database schema or specialized binary formats. Data do not need to be duplicated in different formats, either. Secondly, tabix works on compressed data files while most SQL databases do not. The GenCode annotation GTF can be compressed down to 4%. Thirdly, tabix is fast. The same indexing algorithm is known to work efficiently for an alignment with a few billion short reads. SQL databases probably cannot easily handle data at this scale. Last but not the least, tabix supports remote data retrieval. One can put the data file and the index at an FTP or HTTP server, and other users or even web services will be able to get a slice without downloading the entire file.
What things can you do in bioinformatics?
- Look at variants - which ones matter?
- Assemble genomes for new organisms
- Compare genomes of different organisms
- Biomarker studies:
- Compare gene expression levels across case and control samples, see if you can find significantly different ones. (It's messy. Also, R is better for this than Python (shhh...))
- Molecular interaction networks